Rhawn Joseph, Ph.D.
BrainMind.com
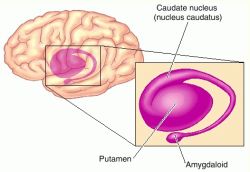
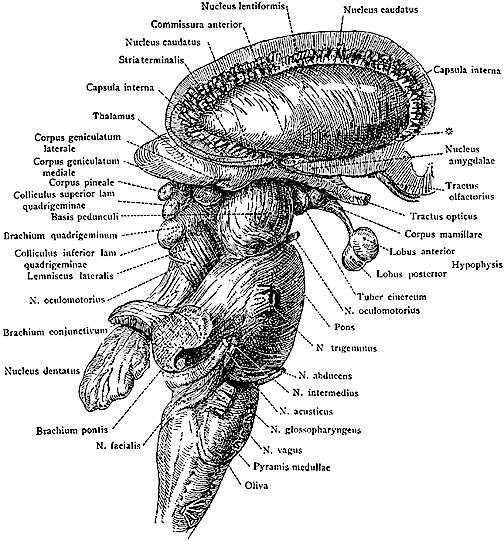
The nucleus accumbens is part of the Basal Ganglia (e.g. caudate, putamen, globus pallidus), and constitutes a major part of the Limbic Striatum. Early in the course of vertebrate evolution, motor functioning was the province of the spinal cord, brainstem, cerebellum, and forebrain (see chapter 5), just as it is in modern species including humans. Much of the forebrain, however, consisted of the olfactory bulbs and tracts, and the amygdalostriatal gray which was both dorsal and ventrally located and contained at its central-posterior-dorsal core, the hippocampus. The amygdalostriatal gray was dominated by the olfactory system, and reacted to olfactory impulses by feeding, fighting, fleeing, or engaging in sexual behavior. Although the amygdalostriatal gray became separate structures once terrestrial vertebrates had evolved (Gloor, 2007; Stephans, 2014; Ulinsky, 2017), the striatum and related structures collectively referred to as the basal ganglia, continued to govern all aspects of affectively triggered gross motor behavior, just as they do in modern humans, albeit in conjunction with the brainstem, cerebellum, spinal cord and cranial nerve nuclei, as well as the thalamus, and the primary, secondary and supplementary motor areas of the frontal lobes.
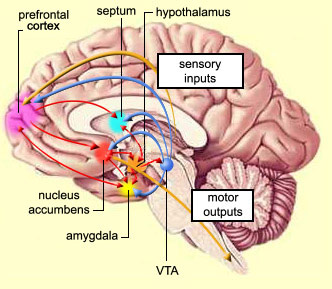
The basal ganglia, brainstem, spinal cord, and frontal motor areas are all interlinked and function as an integrated system in the production of movement. For example, the basal ganglia provides input to the brainstem (via the "extra-pyramidal motor system") as well as to the motor thalamus and motor neocortex which also projects to the brainstem and spinal cord (Parent & Hazrati 1995). If injured, or if abnormalities develop secondary to excessive or diminished levels of neurotransmitters such as dopamine (e.g. Fahn, 2016; Turjanski et al, 2016; Wolters, 2016), a host of cognitive, affective, and motor disturbances may ensue, including Parkinson's and Alzheimer's diseases, chorea, hemiballismus, restless leg syndrome, catatonia, schizophrenia, obsessive-compulsions, or depression (e.g. Aylward et al. 2015; Castellanos et al. 2015; Chakos et al. 2015; Deicken et al. 1995; Ellison 2015; Harrison, 2016; Joseph, 2016a; Turjanski et al, 2016), depending on the nature, degree, extent, and laterality of destruction.
The nucleus accumbens contains cells which selectively respond to motivationally significant stimuli, including novel and familiar variables that are rewarding or punishing (Rolls & Williams, 1987; Schneider & Lidsky, 1981). Striatal neurons can also react differently to familiar stimuli depending on their reinforcement properties, and many neurons will in fact increase their responsiveness as stimuli approach the mouth, and/or touch the face and mouth (Rolls & Williams, 1987; Schneider & Lidsky, 1981). Some striatal neurons also respond when making tongue and lip movements (e.g. licking) and during arm movements toward a food item (Rolls & Williams, 1987).
These findings suggest the striatum is involved in orienting and guiding movements toward the mouth presumably so that a desired object can be licked, sucked on, chewed, and consumed. In this regard the striatum could be considered a primary motor center which enables various limbic desires, needs, and impulses such as hunger, to be satisfied.
The nucleus accumbens and the mesolimbic DA system which projects to other basal ganglia nuclei also appear to be highly involved in mediating feelings of pleasure including the rewarding effects of amphetamine, cocaine and opiates (see Ellison, 2015; Hakan et al. 2015; Koob et al. 2011). These effects (including the aversiveness of opiate withdrawal) are probably due not only to the presence of opiate, DA, and related receptors, but the rich interconnections maintained with the amygdala as well as the lateral hypothalamus (Kelsey & Arnold 2015; Olton et al. 2016; Zaborszky et al. 2011) which constitute part of the "pleasure circuit" maintained by the medial forebrain bundle (Olds & Forbes, 1981; see also chapter 13).
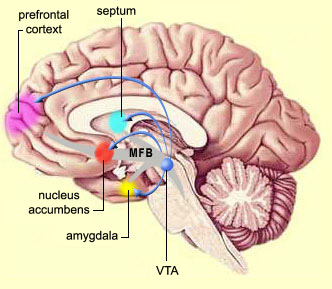
However, animals will also work in order to self-administer opiates directly into the accumbens (Koob et al. 2011; Olds & Forbes 1981). Conversely, lesions to the nucleus accumbens may disrupt the capacity to experience pleasure or the rewarding effects of opiates and cocaine (Koob et al. 2011); or to engage in complex coordinated defensive acts (see below).
Catatonia, Parkinson's Disease, & Psychosis.
Lesions to the corpus striatum and lenticular nucleus (putamen and globus pallidus) can attenuate one's capacity to motorically express their emotions via the musculature; e.g. the face may become frozen and mask-like. These latter motor disturbances are well known symptoms associated with Parkinson's disease, a disturbance directly linked to dopamine deficiency (Fahn, 2016) and neuronal degeneration not only in the putamen (Goto, et al. 2010; Kish, et al. 2018; see also Hauser et al., 2016), but within the limbic striatum, i.e. the nucleus accumbens (see Rolls & Williams, 1987), as well as in the supplementary motor areas and medial frontal lobe -which maintains rich interconnections with the striatum.
Other disturbances associated with striatal abnormalities include Huntington's chorea, ballismus, restless leg syndrome, sensory neglect and apathy, obsessive compulsive disorders, mania, depression, "schizophrenia" and related psychotic states (Aylward et al. 2015; Baxter et al. 2012; Caplan, et al. 1990; Castellanos et al. 2011; Chakos et al. 2012; Davis, 1958; Deicken et al. 1995; Ellison, 2015; Rauch et al. 2017; Richfield, et al. 1987; Turjanski, et al., 2016). Severe memory loss and social-emotional agnosia and an inability to recongize friends or loved ones is also characteristic of striatal abnormalities, particularly disturbances involving the limbic striatum.
Thus although the basal ganglia is often viewed and described as a major motor center (detailed below), the functional capacities and symptoms associated with this group of nuclei are quite diverse, and vary depending on the nuclei and chemical neurotransmitters involved as well as the laterality, location, and extent of any lesion.
THE LIMBIC STRIATUM, DA, 5HT, ACH, NE, & MEMORY
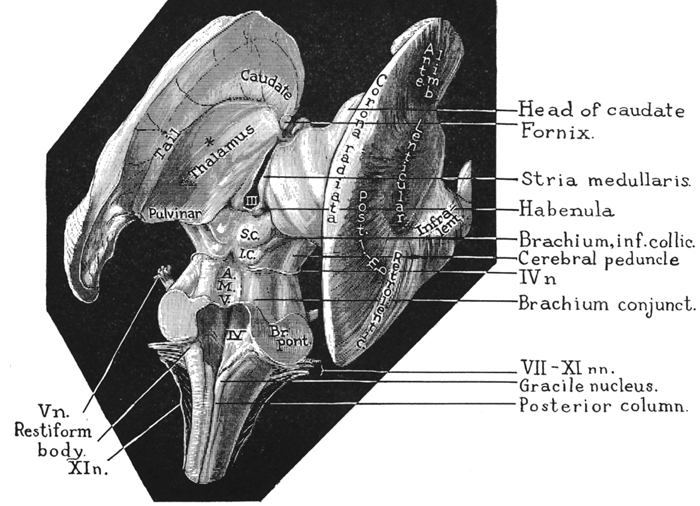
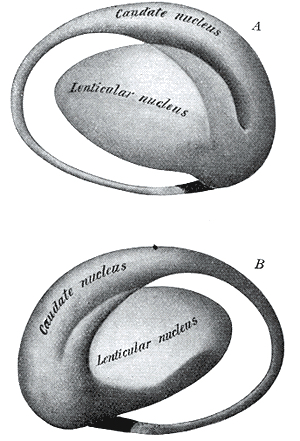
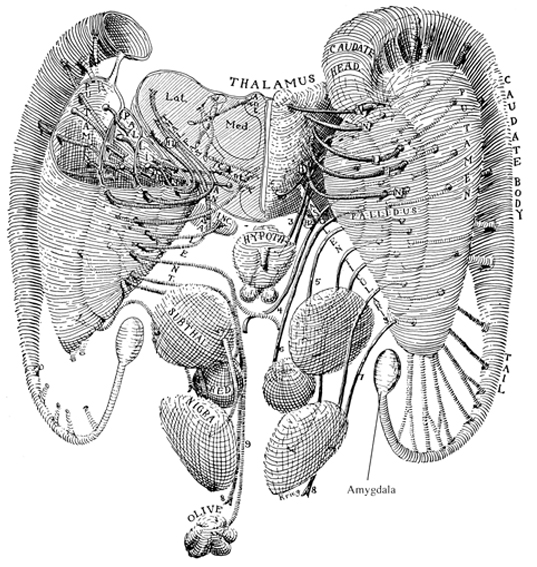
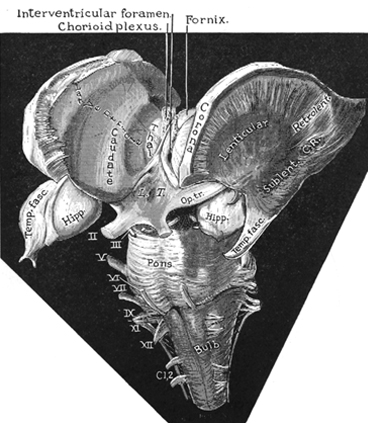
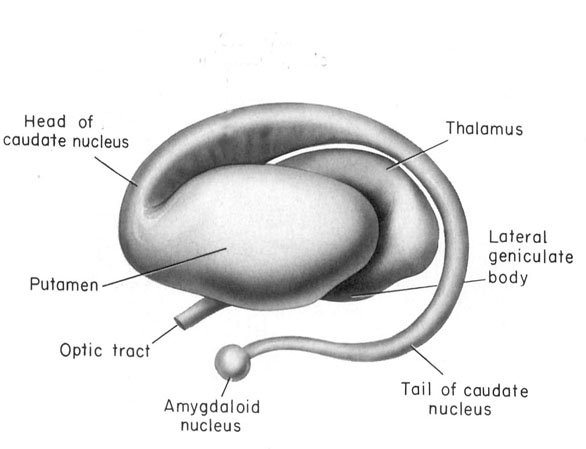
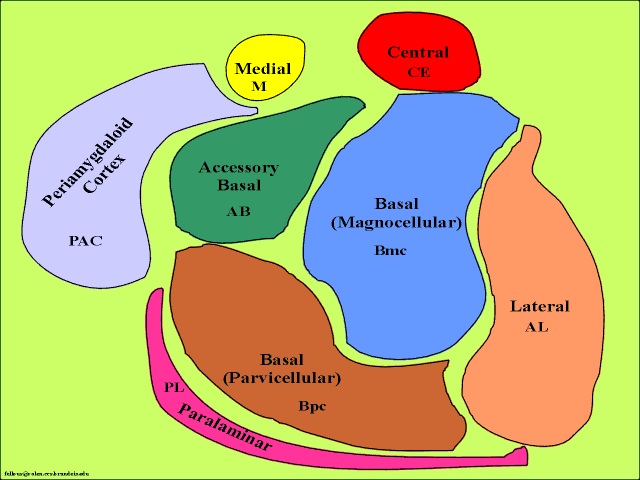
The limbic striatum consists of the nucleus accumbens, olfactory tubericle, the extended (centro-medial) amygdala, as well as the ventral aspects of the caudate, putamen and globus pallidus (-substantia innomminata).
The nucleus accumbens is located immediately beneath the anterior portion of the caudate and from a microscopic level appears to be part of the ventral caudate (Olton et al. 2011; Zaborszky et al. 2011). However, it maintains extensive interconnections with the amygdala as well as the hippocampus via the fimbria-fornix fiber bundle (DeFrance et al. 1985), which implicates this nuclei in memory functioning and probably the learning of visual-spatial and perhaps social-affective relationships.
Similarly, the dorsal portion of the substantia innominata ("great unknown") merges (dorsally) with the ventral globus pallidus (and ventrally with the centro-medial amygdala), and maintains interconnections with the accumbens, hippocampus, lateral amygdala, and dorsal medial (DM) nucleus of the thalamus (Young et al. 1984; Zaborszky et al. 2011) -the DM being involved in regulating neocortical arousal and information reception as well as memory (see chapter 19).
Like the accumbens, the substantia innominata (SI) is also important in memory functioning and has been implicated as one of the principle sites (along with the nucleus basallis) for the initial development of Alzheimers disease (see Olton et al. 2011; Zaborszky et al. 2014).
The nuclei of the limbic striatum are also interconnected with the lateral and medial hypothalamus, brainstem reticular formation, and the frontal and inferior temporal lobes (Everitt & Robbins, 2012; Groenewegen, et al. 2011; Heimer & Alheid, 2001; Kelly, et al. 1982; Mogenson & Yang 2011; Olton et al. 2011; Zaborszky et al. 2011). The limbic striatum is able, therefore, to exert widespread effects on neocortical and subcortical structures.
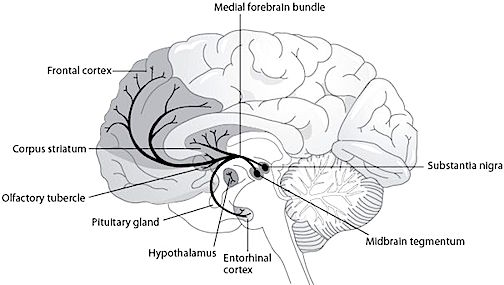
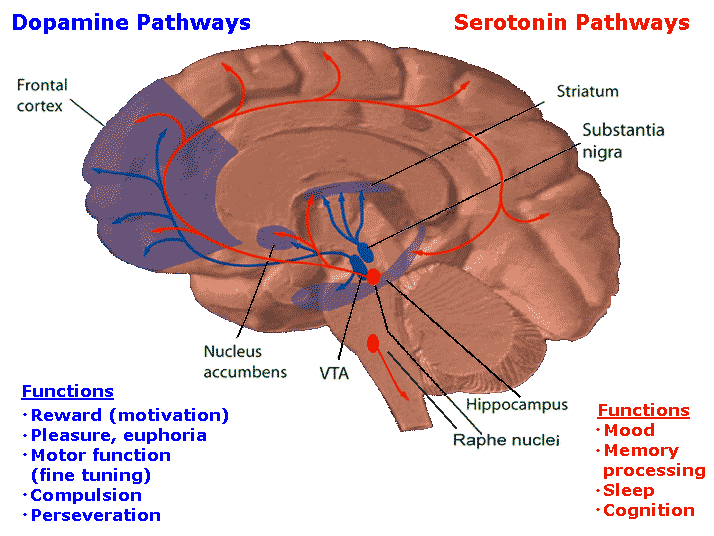
As noted, the limbic striatum receives DA from the mesolimbic system as does the amygdala. The amygdala also sends axons that terminate in striatal neurons immediately adjacent to those innervated by mesolimbic DA neurons (Kelly et al. 1982; Yim & Mogenson, 1983, 1989). Thus, a complex interactional loop is formed between these nuclei, the integrity of which, in part is dependent on the mesolimbic DA system which can act on the amygdala, hippocampus and limbic striatum simultaneously so as to modulate striatal reception of amygdala (Maslowski-Cobuzzi & Napier, 2015) and hippocampal excitatory signals, and regulate the transmission of accumbens input into the SI. Alterations in mesolimbic DA activity, therefore, can significantly influence limbic striatal, amygdala and hippocampal activity as well as the motoric expression of limbic impulses.
For example, depletions in mesolimbic DA can disrupt motor and social-emotional memory functioning; a condition compounded by DA influences on acetylcholine (ACh) neurons, and the effects of striatal DA on the reception of hippocampal, amygala, and neocortical input.
Indeed, the limbic striatum (and limbic system) contain high densities of ACh neurons which are involved in memory as well as motor functioning (Olton et al. 2011; McGaugh et al. 2012; Zaborszky et al. 2011). In fact, of all striatal nuclei, densities of DA (and ACh) neurons are highest within the (SI) and nucleus accumbens (Meredith et al. 1989) which in turn greatly influences the SI (which is a major source of neocortical ACh), as does the amygdala (Yim & Mogenson 1983) and hippocampus; i.e. these signals converge on the SI.
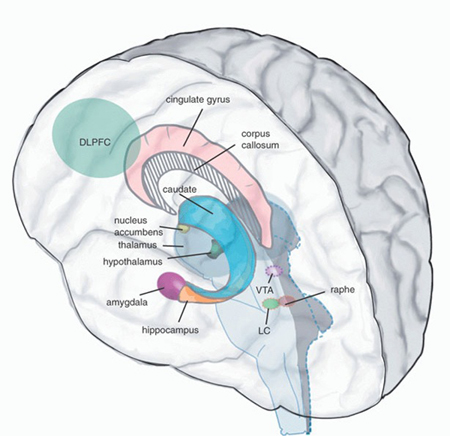
Like the striatum, the hippocampus (and the amygdala) also receives meso-limbic DA input and contains D1 and D2 receptors (Camps et al. 1990) as well as ACh. ACh influences on neuronal activity can be excitory or inhibitory depending on the resting membrain potential of the receiving neuron (Ajima et al. 1990) as well as concurrent DA activity. Specifically, mesolimbic DA mediated signals are relayed from the accumbens to ACh neurons in the SI which in turn project to DM and the neocortex, the frontal lobes in particular (Mogenson & Yang 2011; Olton et al. 2011; Zaborszky et al. 2011). However, the DA and cholinergic, ACh system appear to exert counterbalancing influences.
For example, ACh is usually inhibited by DA. By contrast, DA depletion results increased ACh and neuron hyperactivity (see Aghanjanian & Bunney, 1977; Bloom et al. 1965; Klockgether, et al., 1987) which can greatly disrupt cognitive and memory functioning as well as motor activities, unless reversed by anti-cholinergic drugs. In fact, damage to this DA/cholinergic system can result in Alzheimers disease (Olton et al. 2011; Zaborszky et al. 2011).
As per memory functioning, it appears that mesolimbic DA modulates the reception of hippocampal (motor-spatial) input into the accumbens which projects to the SI (which in turn distributes these influences, perhaps via ACh) to the neocortex). DA accomplishes this via inhibitory influences on ACh and facilitation of the GABA system which appears to exert inhibitory influences within the striatum and the transmission of impulses from the nucleus accumbens (and amygdala/hippocampus) to the SI. Therefore, if the inhibitory influences of GABA and ACh on the SI are dampened, the SI becomes activated and memory functioning may be enhanced or disrupted at the neocortical level as this nucleus exerts widespread ACh influences on the cerebrum. Thus fluctuations in DA levels can either act to excite or inhibit cognitive-memory activity within the limbic striatum (see Mogenson & Yang 2011) as well as the corpus striatum (Packard & White 2011) and the neocortex.
However, also of importance in this memory-striatal neural network is (5HT) serotonin (McLoughlin et al. 2015; Mogenson & Yang 2011) and (NE) norepinephrine (Roozendaal & Cools, 2015). For example, NE levels have been shown to fluctuate within the amygdala and nucleus accumbens when presented with novel stimuli (Cools et al. 2011) and during information acquisition. Presumably NE levels within the accumbens can regulate or influence the reception of amygdala and hippocampal impulses within the accumbens and SI which in turn projects to the neocortex and, in this regard, may act to shunt amygdala-hippocampal impulses to discrete or wide areas of the cerebrum.
Specifically, high alpha-NE activity is associated with reduced amygdala input, whereas low beta NE may reduce hippocampal input into the striatum. Conditions such as these, however, are most likely to result when stressed or traumatized in which case it is possible for amygdala (and thus emotional) input to be received in, learned, and expressed by the striatum in the absence of hippocampal participation (see Rozzendaal & Cools 2015). It is conditions such as these that can give rise to amnesia with preserved learning (see chapters 14, 30).
However, in some cases, it is also possible for both the alpha and beta NE system to be effected simultaneously such that amygdala and hippocampal input to the limbic striatum are inhibited in which case profound memory loss may result (see Rozzendaal & Cools 2015). Similar disturbances are associated with the serotonin (5HT) system (McLoughlin et al. 2015).
Depletion of 5HT can significantly effect the capacity to inhibit irrelevant sensory input (at the level of the brainstem, amygdala, basal ganglia, and neocortex). Hence, 5HT abnormalities or depletion typically results in confusion and sensory overload and in some instances the production of hallucinations (see chapter 30).
Thus disruption of the mesolimbic DA system and/or severe disruptions in the 5HT system in turn interferes with the hippocampal-limbic striatal memory system (Packard & White 2011) and can create neuronal hyperactivity -a condition which interferes with perceptual and hippocampal- memory-SI-amygdala functioning. In this regard it is noteworthy that the central 5HT system is severely disrupted among those with severe memory loss and Alzheimer's disease (McLoughlin et al. 2015) and that calcification of the globus pallidus/SI can induce visual and auditory hallucinations as well as cognitive deterioration (Lauterbach et al. 2015).
OVERVIEW: ALZHEIMER'S DISEASE
Alzhiemer's disease is associated with a profound loss of memory and cognitive and intellectual functioning, including, at its later stages, an inability to recognize friends, loved one's or their own personal identity. Alzhiemer's disease is estimated to afflict approximately 10% of those over age 65, and 50% of those over 85.
In its later stages, Alzhiemer's disease is associated with profound loss of cerebral functional capacity, coupled with neural degeneration and a loss of neurons and the development of amyloid (senile) plaque and neurofibrillary tangles. Because structures such as the substantia innominata, amygdala, and entorhinal cortex have been injured (Morrison et al. 1990; Gomez-Isla, et al., 2000; Rapoport 1990) it is thought that the destruction of this tissue and adjacent tissue accounts for the loss of memory, and social-emotional, facial recognition, and related visual abnormalities, including visual agnosia (Giannakoulos, et al., 2016).
However, because there is no single factor that has been implicated, and due to the number of associated disturbances, it has been argued that Alzheimer's disease and associated cognitive and memory disturbances are due to a "global cortico-cortical disconnection" syndrome (Morrison et al. 1990; Rapoport 2010). Nevertheless, for the purposes of this chapter, the striatal contribution to this disorder will be emphasized.
MEMORY & ALZHEIMER'S DISEASE
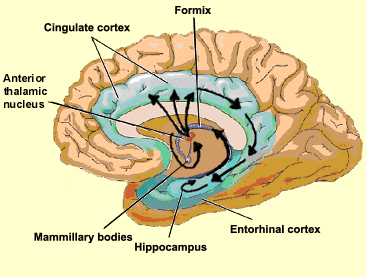
As detailed in chapter 15, the amygdala and hippocampus provide massive input to the limbic striatum as well as the dorsal medial nucleus of the thalamus (DM) , the frontal lobes and reticular activating system, as does the accumbens and SI ( Koob et al. 2001; Mogenson & Yang 2014; Zaborszky et al. 2015). As noted, these nuclei, including the overlying entorhinal cortex, play a significant role in memory and the gating of information destined for the neocortex and appear to be part of a massive neural network designed to control information processing and to establish memory related neural networks (see also chapter 14). Presumably the nucleus accumbens apparently acts to integrate hippocampal and amygdala input, which is then transmitted to the SI which is also the recipient of limbic impulses concerned with cognitive, memory, and motoric activities (see Mogenson & Yang 2011).
In part, it appears that the accumbens and SI play an inhibitory (filtering) role on information processing, and may exert inhibitory (and counterbalancing) influences on the the medial frontal lobes, the DM, and corpus striatum (see Mogenson & Yang 2011). The amygdala exerts similar influences on these nuclei (chapter 15) and is also able to inhibit the SI and accumbens.
However, the role of the limbic striatum in cognitive and memory related activity also includes the learning of reward-related and aversion processes, the facilitation of approach and withdrawal responses and the memorization of where a reward or aversive stimulus was previously received (Everitt et al. 2011; Kelsey & Arnold 2015). In this regard the nucleus accumbens and SI are dependent on the medial and lateral amgydala, especially in the learning of negative experiences (Kelsey & Arnold 2015).
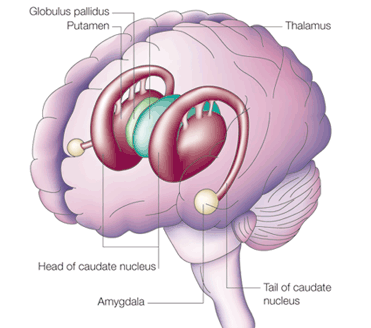
As noted, the SI is the primary source of neocortical cholinergic innervation (Carpenter 2011) and appears to be concerned with integrating social-emotional and cognitive input with motor memories and then storing them perhaps within the SI as well as within the neocortex. Hence, if the SI is lesioned the cholinergic projection system is disrupted, and cognitive as well as social-emotional memory functioning is negatively impacted.
Since the SI merges with and becomes coextensive with the centromedial amygdala (Heimer & Alheid 2011),and is an amygdala derivative, not surprisingly, a significant loss of neurons, neurofibrillary changes and senile plaques have been found in the amygdala (and hippocampus, Rapoport 2010), as well as in the SI, in patients with Alzheimer's disease and those suffering from degenerative disorders and memory loss (Herzog & Kemper, 1980; Mann, 2012; Sarter & Markowitsch, 1985). Degeneration in the amygdala would also account for the social-emotional agnosia and prosopagnosia that is common in the advanced states of Alzhiemer's disease; i.e. failure to recognize or remember loved ones (chapter 13).
WIDESPREAD NEURONAL DEATH & ALZHEIMER'S
Neuronal loss has also been reported in the entorhinal and parahippocampal areas of the inferior temporal lobe (see Morrison et al. 1990; Rapoport 1990) within which is buried the hippocampus and amygdala. Specifically, in addition to senile plaques, neurofibrillary tangles, a 40% to 60% loss of neurons during the early stages of this disease have been found in layers 4 and 2 of the entorhinal cortex (Gomez-Isla, et al., 2000)--the gateway to the hippocampus. Destruction of this tissue and adjacent tissue would also result in memory, social-emotional, facial recognition, and related visual abnormalities, including visual agnosia, as recently demonstrated among those with Alzheimer's disease (Giannakoulos, et al., 2016).
Indeed, among those with Alzheimer's disease, widespread atrophy, amyloid plaques, tangles, and metabolic disturbances have been noted in a variety of cortical areas with relative preservation of the motor and primary receiving areas (Rapoport 1990). Based on these findings it has been argued that Alzheimer's disease and associated cognitive and memory disturbances are due to a "global cortico-cortical disconnection" syndrome (see Morrison et al. 1990; Rapoport 1990); i.e., a loss of neurons in and interconnections with association neocortex, which in part would account for the cognitive deterioration.
The initiation of this deteriorative process may well be in the SI, and it may be due to an olfactory borne infection due to viral or bacterial invasion. That is, these bacterial or viral agents may enter the olfactory system and invade those structures directly innervated by the olfactory nerves, i.e. the amygdala, entorhinal cortex, and SI (Joseph, 1998d). On the other hand, or perhaps related to this scenario, are findings suggesting a genetic foundation for this disorder, such that defects in at least four specific genes (located on chromosomes 1, 14, and 21) may be responsible (Levy-Lehad et al., 1995; Saunders et al., 2013).
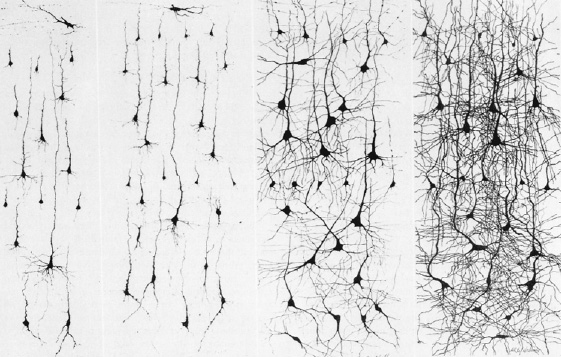
Given the likelihood that the limbic striatum and olfactory-limbic structures may be selectively involved in the early stages of Alzheimers, and given the fact that the SI provides cholinergic input to the neocortex, then the subsequent and progressive loss of SI (and amygdala) neurons might result in a progressive deterioration and cell death within the neocortex such that otherwise healthy neurons are killed. That is, since the SI contains high concentrations of cholinergic neurons which in turn project to widespread areas throughout the neocortex (reviewed in Carpenter 2011), perhaps the initial cell loss within the SI (also referred to as the nucleus basalis), may trigger further cell death in healthy neurons (which project to or receive fibers from the affected cells) which essentially become "pruned" and drop out form disuse.
Dying vs healthy neurons
Defective Axonal Transport.
There is evidence which indicates that perhaps due to head injury, drug or toxic exposure, or perhaps the loss of synaptic junctions (due to the death of unhealthy cells and dendritic retraction), axonal transport becomes defective due to the death of its target neuron. However, if a healthy cell cannot discharge and exchange information it too may die, thus leading to a domino effect and thus widespread cell death (see Burke et al. 2012). That is, due to the loss of terminal synaptic junctions (due to cell death) or to other chemical abnormalities including defects in microtubule assembly (which participates in neuronal transmission), axonal transport becomes dysfunctional as the receiving cell and it's dendrite have died. Hence, there is a buildup of toxic oxidatative metabolites and naturally occurring neurotoxins in the healthy cell body that project to the dead cell, which causes the normal cell to die as well. This would result in a progressive loss of neurons such that widespread areas of the cerebrum soon become effected.
Presumably this what may occur if the limbic striatum becomes abnormal and cells within the SI and accumbens begin to die. As the disturbance and deterioration spreads, cognitive, emotional, memory and related abnormalities including Parkinsonian symptoms begin to appear and become progressively worse as neocortical, striatal, and limbic neurons die and drop out.
LIMBIC & CORPUS STRIATAL COUNTERBALANCING INFLUENCES
The limbic and corpus striatum are richly interconnected and send projections to many of the same brain areas. However, due to differential input from the amygdala and DA systems, these nuclei exert tremendous counterbalancing influences on each other and their associated neural networks. For example, the centro-medial as well as the basolateral amygdala projects to and exerts excitatory influences on the the limbic striatum (Maslowski-Cobuzzi & Napier, 2015; Yim & Mogenson 1982), whereas the medial and posterior-lateral amygdala projects to the corpus striatum and the ventral and dorsal globus pallidus. Via these dual interconnections, the amygdala can exert simultaneous and even oppositional influences on these nuclei.
In addition, the corpus striatum receives the bulk of its DA from the nigrostriatal systems, whereas the limbic striatum receives DA from the mesolimbic DA system (Fibiger & Phillips, 2016) -transmitter systems which interact at the level of the brainstem, limbic system, striatum, and neocortex (Maslowski-Cobuzzi & Napier, 2015). For example, it has been shown mesolimbic DA can act on the amygdala and limbic striatum simultaneously so as to modulate striatal reception of amygdala excitatory signals (Maslowski-Cobuzzi & Napier, 2015).
However, because the corpus and limbic striatum are largely (but not completely) innervated by different clusters of midbrain DA neurons, reductions or increases in one DA system can exert profound influences on those neurons innervated by the others -for example, by eliminating inhibitory or counterbalancing influences. Thus depletion of DA in the nigrostriatal (but not the mesolimbic) pathways can result in increased activity within the limbic striatum (see Yim & Mogenson, 1983, 1989) and medial amygdala, but deceased activity within the corpus striatum. If this occurs, movement programming may be disrupted resulting in rigidity or tremors -a consequence, in part, of an imbalance in amygdala-DA-striatal activation.
Conversely, mesolimbic DA depletion can result in enhanced corpus striatal activity and decreased limbic striatal and lateral amygdala and hippocampal activity, such that motor and social-emotional memory functioning may be disrupted; a condition compounded by DA influences on acetylcholine (ACh) neurons and the reception of hippocampal, amygala, and neocortical input within the striatum. Hence, the functioning of the limbic or corpus striatum can be severely disrupted even when the functional integrity of its own neurotransmitter systems are otherwise intact; i.e. due to a loss of counterbalancing influences.
REFERENCES